|
|
|
|
|||
|
|
Demyelinating Diseases
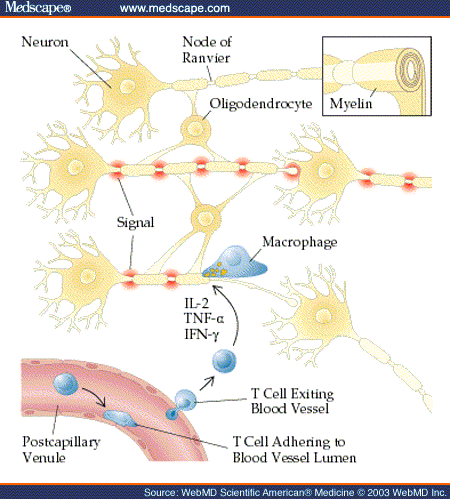
Definition In both the central and the peripheral nervous systems, large-diameter axons are myelinated. Myelin is formed and maintained by oligodendrocytes within the central nervous system and by Schwann cells in the peripheral nervous system. Myelin both insulates the invested axons and organizes surface membrane constituents of the axon--functions that are critical for the rapid transfer of signals necessary for coordinated motor activity, proper integration and interpretation of sensory stimuli, and facile cognition. Diseases that affect the integrity of the oligodendrocyte and its ability to produce and maintain myelin or diseases that directly damage the myelin sheath disturb conduction in myelinated white matter pathways and produce a broad array of motor, sensory, and cognitive dysfunctions [see Figure 1].
Demyelinating diseases disturb the integrity of myelin, but the axons are relatively spared [see Table 1]. These diseases primarily affect oligodendroglial survival (e.g., progressive multifocal leukoencephalopathy), oligodendroglial metabolism (e.g., vitamin B12 deficiency), and the myelin sheath, with secondary effects on the oligodendrocytes (e.g., multiple sclerosis [MS]). Immune-mediated demyelinating diseases include recurrent or chronically progressive demyelinating diseases (MS and its variants) and monophasic demyelinating diseases (optic neuritis, acute disseminated encephalomyelitis, and transverse myelitis). Monophasic demyelination may be the first clinical sign of MS.
Multiple SclerosisMS is characterized clinically by recurrent or chronically progressive neurologic dysfunction caused by lesions in the CNS. Pathologically, the lesions are multiple areas of demyelination that affect the brain, optic nerves, and spinal cord. Full detail on MS can be found here http://www.medscape.com/viewarticle/453515_2
Optic NeuritisOptic neuritis is an acute inflammatory optic neuropathy. The cardinal symptoms are unilateral vision loss and retrobulbar pain with eye movement. Differential diagnosis includes anterior ischemic optic neuropathy, which is usually painless and found in patients older than 50 years; hereditary diseases, such as Leber hereditary optic neuropathy; and toxic or nutritional optic neuropathies. Treatment with intravenous methylprednisolone at a dosage of 1 g/day for 3 days followed by oral prednisone for 11 days hastens recovery of vision but has little residual benefit at 1 year. One study showed that prednisone at 1 mg/kg/day for 14 days had no benefit and was associated with an excess of recurrences. Even without treatment, almost all patients begin to recover vision within 4 weeks. The relation of optic neuritis to MS is controversial. Some regard optic neuritis as a distinct entity, but others consider it part of the clinical continuum of MS. More than half of all patients with MS have optic neuritis at some time during the disease. Of patients who present with optic neuritis and who have no other neurologic deficit, almost 40% have one or more ovoid or periventricular lesions, as revealed on brain MRI; clinically definite MS eventually develops in 60%. Patients with completely normal results on MRI and comprehensive CSF evaluation seldom progress to MS.
Acute Disseminated EncephalomyelitisAcute disseminated encephalomyelitis (ADEM) is a monophasic syndrome that is usually preceded by a viral exanthem, an upper respiratory infection, or vaccination. The most commonly associated viruses are measles, paramyxovirus, varicella, rubella, and Epstein-Barr virus. Onset is often rapid and is characterized by meningeal signs, headache, seizures, and altered mental status. The associated neurologic deficits are variable and may include hemiplegia, paraplegia, sensory loss, vision loss, and transverse myelitis. ADEM can be fatal, but most patients begin to recover within 2 to 4 weeks. Acute hemorrhagic encephalomyelitis is probably a fulminant variant of ADEM. The main pathologic features of ADEM are multiple areas of perivascular inflammation and demyelination, without evidence of active viral infection. ADEM may be caused by an autoimmune response against myelin antigens elicited by cross-reactive viral proteins. Usually, multiple white matter lesions are present on MRI, and the majority of the lesions can be enhanced with contrast. Corticosteroid treatment is often used, although the efficacy of this approach has not been proved in clinical trials. Plasmapheresis may also be useful.[66] Prognosis varies with the inciting virus, but mortality may be as high as 30%, and survivors may be left with residual symptoms.
Transverse MyelitisAcute transverse myelitis is a syndrome of spinal cord dysfunction; it has a rapid onset. Like ADEM, it may occur after infection or vaccination or it may occur with no discernible precipitant. It may also be the initial presentation of MS. Symptoms include paraparesis, which is initially flaccid and then spastic; loss of sensation with a sensory level on the trunk; and bowel and bladder dysfunction. Back pain precedes the neurologic symptoms, and the sensory symptoms may begin distally and ascend. The thoracic cord is most often affected. The differential diagnosis includes other causes of acute myelopathy, such as compression of the cord by an extradural structural lesion, spinal cord neoplasms, ischemia, and systemic lupus erythematosus. MRI is extremely useful for excluding structural lesions and for confirming the presence of an intramedullary lesion at the level in the spinal cord commensurate with the symptoms. The lesions of acute transverse myelitis are typically hyperintense on T2-weighted imaging; they involve the majority of the cross-sectional area of the cord over several segments and may be enhanced with contrast. The lesions may cause swelling of the spinal cord. No treatment has proved beneficial, but corticosteroids are often used. Prognosis is variable: one third of patients have a good outcome, one third have a fair outcome, and one third do not recover. Spinal shock, back pain, and catastrophic onset are associated with poor outcome.
Inherited Demyelinating DiseasesAdrenoleukodystrophy is an inherited disorder that is associated with progressive demyelination and dysfunction of the adrenal cortex. The inheritance pattern may be either autosomal recessive or X-linked recessive. The X-linked form is caused by the mutation of a gene encoding an integral membrane protein found in the peroxisome. Defects in this gene lead to accumulation of very long chain fatty acids (VLCFAs). The phenotypes may vary considerably, even within the same family. In the childhood form, the patient presents with cognitive deficits; rapid neurologic deterioration then occurs, with death occurring in 2 to 5 years. The adult form, called adrenomyeloneuropathy, presents at a mean age of 28 years as progressive spinal cord dysfunction with spastic paraparesis, sensory loss, and bowel and bladder symptoms. Cerebral involvement may be minimal. Only half of patients with adult-onset disease have brain abnormalities on MRI; these are most often found in the corticospinal tracts. Most patients have diffuse atrophy of the spinal cord. Diagnosis is made on the basis of the combination of neurologic and adrenal involvement, family history, and elevated levels of serum VLCFAs. Dietary treatment with unsaturated fatty acids lowers the level of VLCFAs but does not significantly affect the progression of symptoms. Bone marrow transplantation may be effective if performed before severe symptoms develop. Prognosis is poor for patients with the childhood form of disease. Patients with adult-onset disease usually require assistance with ambulation within 10 to 15 years, and rapidly progressive cerebral lesions develop in a large percentage of patients 5 to 10 years after the onset of spinal cord symptoms. Metachromatic leukodystrophy is an autosomal recessive disorder that results in demyelination of axons in the central and peripheral nervous systems. It is caused by mutations in the gene for arylsulfatase A that lead to accumulation of metachromatically staining sulfatides. Onset usually occurs in infancy or childhood; adult onset is rare. The symptoms of adult-onset disease are progressive behavioral abnormalities, dementia, ataxia, and neuropathy. MRI or CT of the brain demonstrates atrophy and diffuse white matter abnormalities, particularly in the frontal lobes. Diagnosis is confirmed by measurement of arylsulfatase A activity in peripheral blood leukocytes, urine, or skin fibroblasts. True arylsulfatase deficiency must be distinguished from a common pseudodeficiency state that is caused by an allele with low enzymatic activity. The symptoms of metachromatic leukodystrophy are relentlessly progressive, and earlier onset is associated with more rapid progression. The mean survival for adult-onset disease is about 12 years. No effective treatment is available, but allogeneic bone marrow transplantation and gene therapy are under investigation.
Metabolic Demyelinating DiseasesCentral pontine myelinolysis (CPM) is a syndrome in which neurologic deficits occur after rapid correction of hyponatremia. CPM usually occurs in young to middle-aged adults and is often associated with alcohol abuse or malnutrition. Signs and symptoms usually begin 3 days after the start of sodium replacement and consist of changes in mental status, dysarthria and other signs of corticobulbar dysfunction, and spastic quadriplegia. Improvement usually begins about 2 weeks after the onset of symptoms, but the degree of recovery is variable. The most striking finding on pathologic examination is the presence of symmetrical demyelinated lesions in the central pons. Demyelinated lesions may also occur in a relatively symmetrical pattern in the basal ganglia, thalamus, internal capsule, subcortical white matter, and cerebellum. T2-weighted MRI usually demonstrates the presence of hyperintense lesions. These lesions usually cannot be enhanced with contrast. CPM may also occur after liver transplantation. There is no specific treatment once symptoms have developed. Long duration and rapid correction of hyponatremia increase the risk of CPM; the recommended rate for correction of hyponatremia is no faster than 10 to 12 mEq in 24 hours. Vitamin B12 deficiency results in demyelination of axons in the central and peripheral nervous systems. The dorsal and lateral white matter tracts of the spinal cord are most affected--a characteristic that has given rise to the name subacute combined degeneration of the spinal cord. The most common presenting symptoms are paresthesias, sensory loss that begins in the feet and progresses proximally, and sensory ataxia. Weakness almost always begins after sensory loss. Memory difficulties, irritability, and confusion occur in a minority of patients. On examination, patients usually have decreased vibration and position sense, which is worse in the feet than in the hands, and may have spastic paraparesis. Pathologic examination reveals symmetrical loss of myelin in the posterior and lateral columns of the spinal cord and sometimes patchy demyelination in the cerebral white matter. MRI of the spinal cord often demonstrates white matter lesions, which resolve with treatment. Diagnosis is made on the basis of the clinical findings and a low serum cobalamin level. Macrocytosis or anemia is present in most patients but cannot be used in place of the cobalamin level as a diagnostic measure. For patients who have symptoms and a low-normal cobalamin level, demonstration of elevated levels of serum methylmalonic acid and total homocysteine can confirm the presence of a functionally significant cobalamin deficiency. If cobalamin deficiency is present, the underlying etiology should be investigated. About 80% of patients with cobalamin deficiency have pernicious anemia. Administration of cobalamin prevents progression of symptoms and produces clinical improvement in most patients. Nitrous oxide prevents the metabolism of cobalamin and can cause similar symptoms after prolonged exposure; after a single exposure, it can unmask a subclinical cobalamin deficiency.
Virus-Induced DemyelinationProgressive multifocal encephalopathy is a lethal demyelinating disease caused by an opportunistic viral infection of oligodendrocytes in immunocompromised patients. The causative agent is JC virus, a ubiquitous papovavirus that infects the majority of the population before adulthood and establishes a latent infection in the kidney. In immunocompromised hosts, the virus can reactivate and productively infect oligodendrocytes. This previously rare condition is now more common because it occurs in 4% of patients with AIDS. Patients usually present with relentlessly progressive focal neurologic deficits, such as hemiparesis or visual field deficits, or with alterations in mental status. On brain MRI, one or more white matter lesions are present; they are hyperintense on T2-weighted images and hypointense on T1-weighted images. There is no mass effect, and contrast enhancement is rare. Diagnosis can be confirmed by brain biopsy, with demonstration of virus by in situ hybridization or immunocytochemistry. Polymerase chain reaction amplification of JC virus sequences from the CSF can confirm diagnosis without the need for biopsy.[80] Currently, there is no effective therapy. Survival after diagnosis is about 3 to 5 months in AIDS patients. Subacute sclerosing panencephalitis (SSPE) is a rare late complication of measles virus infection. It occurs most often in patients who had the initial infection with measles virus before 2 years of age; the mean lag between initial infection and SSPE is 7 years. The use of measles vaccine has greatly reduced the incidence of this complication in developed countries. The earliest symptom is usually progressive cognitive deterioration, which is followed by motor dysfunction and myoclonus associated with distinctive electroencephalographic abnormalities. Pathologic examination reveals active viral infection in the brain, with measles virus protein and RNA detectable in both oligodendrocytes and neurons, and a vigorous inflammatory response. The course is progressive, with occasional temporary remissions. There is no satisfactory treatment. Page forms part of www.apls.tk, the information site on ANTIPHOSPHOLIPID SYNDROME (APS or ANTIPHOSPHOLIPID SYNDROME (APLS))
|
|